Lecture 9: First Law Revisited
This lecture provides a bridge from our recent discussions on real gases and phase diagrams back to the foundational principles of classical thermodynamics. Our aim is to make the First Law of Thermodynamics operational, clearly distinguishing between state functions and processes. We'll learn how to calculate changes in internal energy for an ideal gas and lay the groundwork for understanding adiabatic relations, which we'll complete in the next session.
By the end of this lecture, you should be able to:
- Calculate changes in internal energy from considerations of work and heat.
- Calculate heat capacities and temperature differences, and understand their connection.
- Understand the consequences of the Joule free expansion experiment.
- Connect microscopic and macroscopic interpretations of the First Law.
- Set up the initial properties for an adiabatic expansion, leading to the full $PV^\gamma$ result next time.

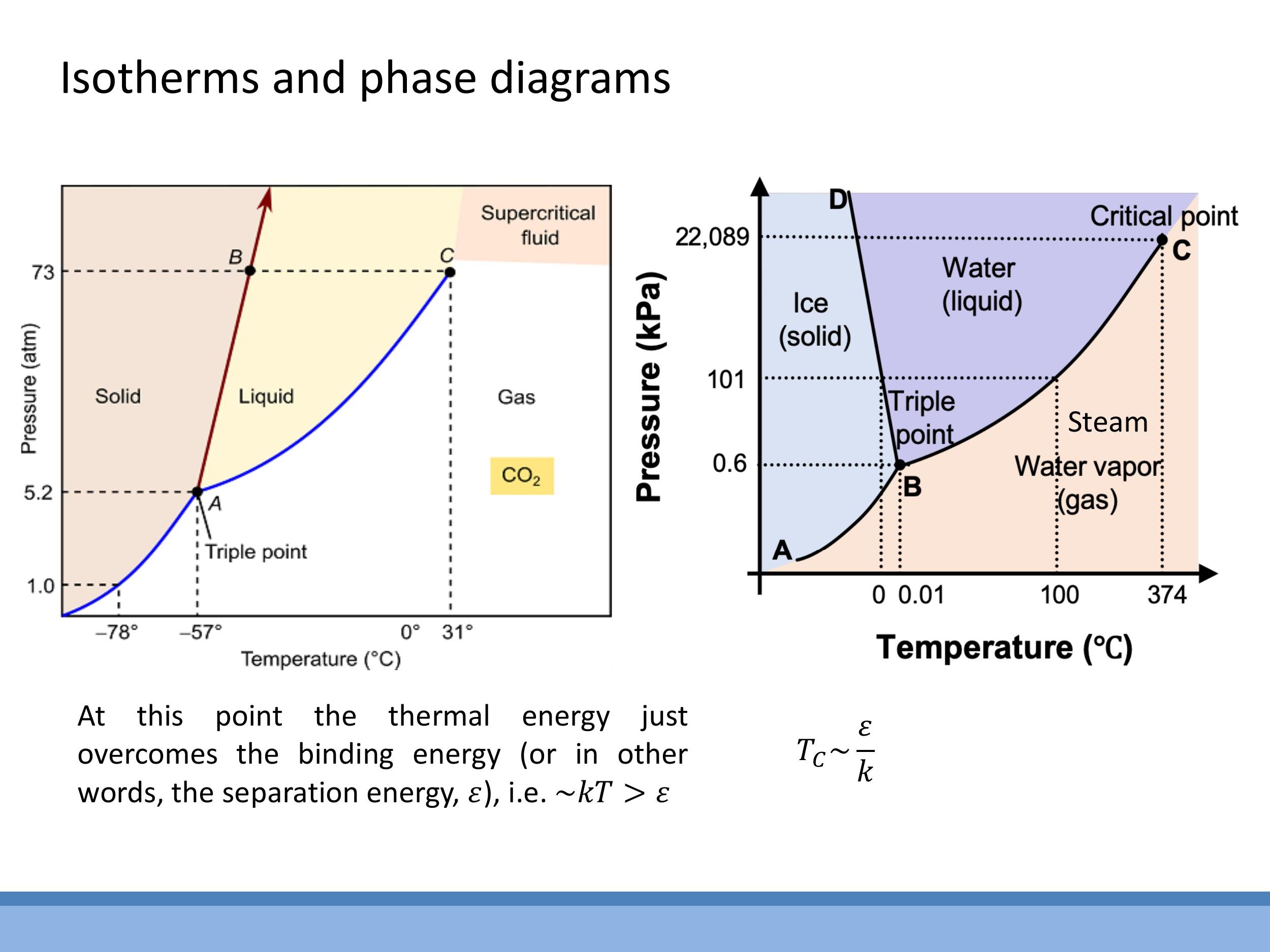
1) Recap: real-gas isotherms, the phase dome, and $T_C \sim \varepsilon/k$
We've previously explored how real gases deviate from the ideal gas law, $PV = RT$. For an ideal gas, an isotherm (a curve of constant temperature on a pressure-volume diagram) is a smooth hyperbola. However, real gases, particularly at low temperatures, exhibit more complex behaviour. Their isotherms can enter a two-phase region, showing a characteristic "loop" that physically represents the process of condensation, where gas and liquid coexist.
The failure of the ideal gas model stems from two main assumptions. Firstly, ideal gases are assumed to have no intermolecular forces, but in reality, attractive forces exist between molecules. These attractions reduce the measured pressure on the container walls, which we account for with the van der Waals $a/V^2$ term. Secondly, ideal gas particles are treated as point-like, having negligible size. Real molecules, however, occupy a finite volume, reducing the free space available for other molecules to move in. This is corrected by the van der Waals $nb$ term, where $b$ represents the excluded volume per mole.
As we increase the temperature, the distinctive "loop" on the real gas isotherm shrinks. At a specific temperature, known as the critical temperature ($T_C$), this loop vanishes, replaced by a single point of inflection. Above $T_C$, no amount of compression can liquefy the gas; the liquid and gas phases become indistinguishable, forming a supercritical fluid. Conceptually, at this critical point, the thermal energy $kT_C$ is just sufficient to overcome the intermolecular binding energy, $\varepsilon$. This provides a useful approximation for the critical temperature: $T_C \sim \frac{\varepsilon}{k}$. This link connects the macroscopic critical temperature to the microscopic bond strength, $\varepsilon$, which we inferred earlier from latent heats and neighbour counting.
Water exhibits an interesting anomaly in its phase diagram. Unlike most substances, its solid-liquid boundary line has a negative slope. This occurs because ice is less dense than liquid water. Consequently, increasing the pressure on ice can cause it to melt, which is contrary to the behaviour of most materials where increased pressure favours the denser solid phase.
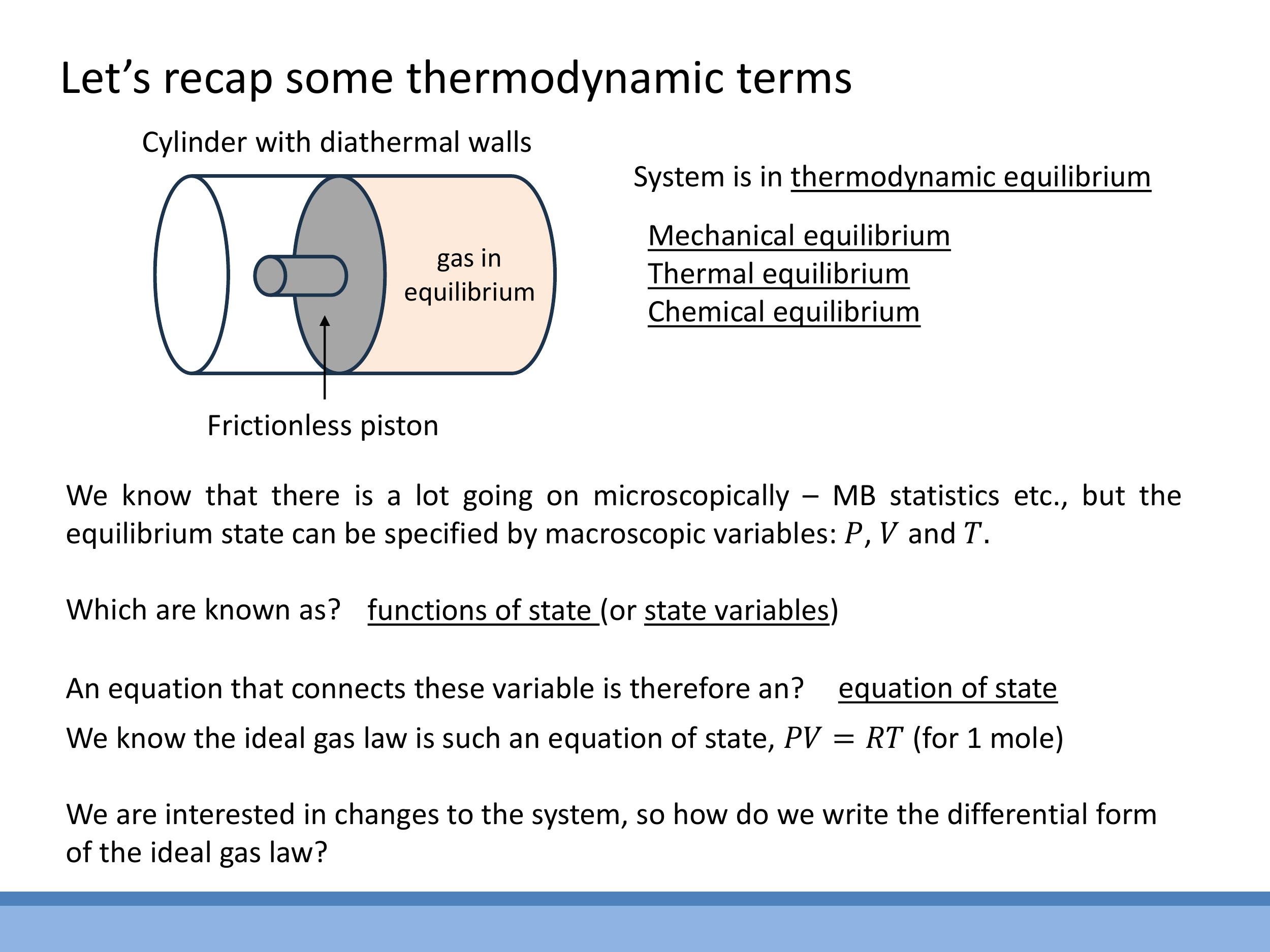
2) Thermodynamic terms and state functions
To understand energy transformations, we often consider a model system: a gas confined in a cylinder with a frictionless piston. The walls of the cylinder can be either diathermal, allowing heat to flow through, or adiabatic, preventing any heat exchange. When such a system is in thermodynamic equilibrium, it means no macroscopic properties are changing. This encompasses mechanical equilibrium (no net forces, so the piston isn't moving), thermal equilibrium (no net heat flow), and chemical equilibrium (no net change in chemical composition).
The macroscopic state of a system is defined by its state variables, or state functions, such as pressure ($P$), volume ($V$), and temperature ($T$). The value of a state function depends only on the current state of the system, not on the path taken to reach that state. Any equation that relates these state variables is called an equation of state; the ideal gas law, $PV = RT$ (for one mole), is a familiar example.
When analysing small changes in a system, it's useful to express the ideal gas law in its differential form. Starting with $PV = RT$ for one mole of an ideal gas, we can consider an infinitesimal change. Applying the product rule for differentiation to the left side, $d(PV) = P \, dV + V \, dP $. The right side simply becomes $ d(RT) = R \, dT$. Equating these, we obtain:
$$
R\,dT = V\,dP + P\,dV
$$
This identity is a cornerstone for calculations involving thermodynamic processes. It directly links small changes in temperature to the corresponding small changes in pressure and volume, allowing us to track how these state variables evolve during a process.

4) Internal energy $U$: what it is and what we can measure
Internal energy, denoted by $U$, represents the sum of all microscopic energies within a system. This includes the translational, rotational, and vibrational kinetic energies of its molecules, as well as the potential energies arising from intermolecular interactions (which are significant in real gases).
Crucially, $U$ is a state function, just like pressure, volume, and temperature. This means its value depends solely on the system's current state, not on the particular path or process used to reach that state. In contrast, heat ($Q$) and work ($W$) are not properties of a system's state; they are processes, representing energy in transit as it moves into or out of the system. Because $U$ is a state function, the change in internal energy, $\Delta U$, between any two states depends only on the initial and final states, making it path-independent. In practical terms, we can only measure changes in internal energy, $\Delta U$, rather than its absolute value. This path independence is formally expressed by the concept of an exact differential, meaning that $dU$ can be written as a function of state variables, such as $dU(V,P) = (\frac{\partial U}{\partial V})_P dV + (\frac{\partial U}{\partial P})_V dP$, and its integral is independent of the path taken.

The First Law of Thermodynamics is a statement of the conservation of energy. It quantifies how the internal energy of a system changes when energy is transferred as heat or work. One common form of the law states that the change in internal energy ($\Delta U$) is the sum of the heat added to the system ($Q$) and the work done on the system ($W$):
$$
\Delta U = Q + W
$$
In this course, for derivations involving infinitesimal changes, we will primarily use the differential form:
$$
dQ = dU + P\,dV
$$
Here, $P \, dV$ represents the infinitesimal work done by the system during an expansion. Thus, this equation states that the heat added to a system ($dQ$) can either increase its internal energy ($dU$) or be used by the system to do work on its surroundings ($P \, dV$). It's important to be aware that different textbooks may use different sign conventions for heat and work. Always check the specific form being used. In our convention, work done by the system enters as a positive $P \, dV$ term on the right-hand side of the equation.
6) Worked example: $\Delta U$ for heating 1 mol monatomic ideal gas from 0°C to 100°C
Let's calculate the change in internal energy when one mole of a monatomic ideal gas is heated from $0^\circ\text{C}$ to $100^\circ\text{C}$. For a monatomic ideal gas, the internal energy $U$ arises solely from translational kinetic energy. For one mole, this is given by $U = \frac{3}{2}RT$. This means that for an ideal gas, the internal energy depends only on its temperature, $T$.
To find the change in internal energy, $\Delta U$, we first find the rate of change of $U$ with respect to $T$:
$$
\frac{dU}{dT} = \frac{3}{2}R
$$
Now, to find the total change in internal energy over a temperature range $\Delta T$, we can separate variables and integrate:
$$
\int_{U_1}^{U_2} dU = \int_{T_1}^{T_2} \frac{3}{2}R\,dT
$$
$$
\Delta U = \frac{3}{2}R\,\Delta T
$$
Given that $\Delta T = 100 \, \text{K} $ (from $ 0^\circ\text{C} = 273.15 \, \text{K} $ to $ 100^\circ\text{C} = 373.15 \, \text{K} $), and using the ideal gas constant $ R \approx 8.314 \, \text{J mol}^{-1}\text{K}^{-1}$:
$$
\Delta U = \frac{3}{2} \times 8.314\,\text{J mol}^{-1}\text{K}^{-1} \times 100\,\text{K} \approx 1.25 \times 10^3\,\text{J}
$$
So, heating one mole of a monatomic ideal gas by $100 \, \text{K} $ increases its internal energy by approximately $ 1250 \, \text{J}$.

7) Joule’s experimental foundations: heat-work equivalence
James Prescott Joule conducted seminal experiments in the mid-19th century that established the quantitative equivalence between heat and mechanical work, laying a crucial foundation for the First Law of Thermodynamics.
In his famous paddle-wheel experiment, Joule used a falling weight to rotate a paddle wheel submerged in an insulated container of water. He measured the mechanical work done by the falling weight and observed a corresponding rise in the water's temperature. He then demonstrated that the same temperature increase could be produced by supplying an equivalent amount of heat. This direct, quantitative equivalence between mechanical work and heat provided strong experimental support for the idea that internal energy can be changed by either process, as captured by the First Law, $\Delta U = Q + W$.

Joule even took thermometers on his honeymoon to the Swiss Alps, where he attempted to measure the temperature change of water falling over a $450 \, \text{m}$ waterfall. The principle is that the gravitational potential energy lost by the falling water is converted into internal energy, causing a temperature rise. We can estimate this temperature change by equating the potential energy to the thermal energy:
$$
mgh = mc_w\Delta T
$$
where $m$ is the mass of water, $g$ is the acceleration due to gravity, $h$ is the height of the waterfall, and $c_w$ is the specific heat capacity of water. Rearranging to solve for $\Delta T$:
$$
\Delta T = \frac{gh}{c_w}
$$
Using $g \approx 9.8 \, \text{m s}^{-2} $, $ h = 450 \, \text{m} $, and $ c_w \approx 4190 \, \text{J kg}^{-1}\text{K}^{-1}$, we find:
$$
\Delta T = \frac{9.8\,\text{m s}^{-2} \times 450\,\text{m}}{4190\,\text{J kg}^{-1}\text{K}^{-1}} \approx 1.05\,\text{K}
$$
Thus, a $450 \, \text{m} $ waterfall would cause the water to warm by approximately $ 1^\circ\text{C}$. This back-of-the-envelope calculation illustrates the magnitude of the effect Joule was trying to measure.

8) Joule free expansion: what $U$ depends on for an ideal gas
Joule also performed a free expansion experiment to investigate how the internal energy of a gas depends on its volume. In this setup, a thermally insulated container is divided into two compartments by a valve. One compartment contains a gas, while the other is evacuated (a vacuum). When the valve is opened, the gas rapidly expands to fill both compartments.
For an ideal gas undergoing this process, no heat is exchanged with the surroundings because the container is thermally insulated ($Q = 0$). Furthermore, since the gas expands into a vacuum, it does no work against any external pressure ($W = 0$). According to the First Law of Thermodynamics, $\Delta U = Q + W$, which means $\Delta U = 0$ for this experiment. Experimentally, for gases that behave ideally, no temperature change ($\Delta T = 0$) is observed during a free expansion. Since the pressure ($P$) and volume ($V$) of the gas change, but its internal energy ($U$) and temperature ($T$) do not, this experiment demonstrates a crucial property of ideal gases: their internal energy $U$ depends only on temperature ($U = U(T)$), not on volume or pressure.
For a real gas, a small cooling effect is actually observed. This is because real gas molecules have attractive intermolecular forces. As the gas expands, its molecules move further apart, doing work against these attractive forces. This internal work consumes some of the gas's kinetic energy, leading to a slight decrease in its temperature.

9) Setting up adiabatic process relations (derivation to be completed next lecture)
We can now begin to set up the relationships for an ideal gas undergoing an adiabatic process, where no heat is exchanged with the surroundings. This derivation will be completed in the next lecture, but we can establish the initial steps.
We start with the First Law of Thermodynamics in its differential form:
$$
dQ = dU + P\,dV
$$
For an adiabatic process, $dQ = 0$. For an ideal gas, we know that the change in internal energy is given by $dU = C_V \, dT $, where $ C_V$ is the molar heat capacity at constant volume. Substituting these into the First Law:
$$
0 = C_V\,dT + P\,dV
$$
This can be rearranged to:
$$
C_V\,dT = -P\,dV
$$
We also use the differential form of the ideal gas law for one mole, $R \, dT = V \, dP + P \, dV $. From this, we can derive Mayer's relation. At constant pressure ($ dP=0 $), the ideal gas law simplifies to $ R \, dT = P \, dV $. Substituting this into the First Law (at constant pressure, $ dQ = C_P \, dT $ and $ dU = C_V \, dT$):
$$
C_P\,dT = C_V\,dT + R\,dT
$$
Dividing by $dT$, we get Mayer's relation:
$$
C_P = C_V + R
$$
These ingredients, including $C_V \, dT = -P \, dV $ and Mayer's relation, allow us to derive the full adiabatic relations linking pressure, volume, and temperature for ideal gases, such as $ PV^\gamma = \text{constant} $ (where $ \gamma = C_P/C_V$). However, the complete derivation of these results will be covered in the next lecture.

Slides present but not covered this lecture (for clarity)
Please note that while the slide deck for this lecture may contain the final $PV^\gamma$ result and its integration steps (often boxed on a slide), this specific content was not covered in this session. The full derivation of these adiabatic relations will be completed in the next lecture.
Key takeaways
Real gases deviate from the ideal gas law ($PV = RT$) because their molecules have finite size and experience intermolecular attractions. These effects give rise to the two-phase "dome" on P-V diagrams and the concept of a critical point, where the distinction between liquid and gas vanishes. The critical temperature ($T_C$) is physically linked to the microscopic intermolecular separation energy ($\varepsilon$) by the approximate relationship $kT_C \sim \varepsilon$.
A system's macroscopic state is defined by state variables like pressure ($P$), volume ($V$), and temperature ($T$). Internal energy ($U$) is also a state function, meaning its value depends only on the system's current state. In contrast, heat ($Q$) and work ($W$) are processes, representing energy in transit, not properties of the state itself.
For infinitesimal changes in an ideal gas, the differential form of the ideal gas law is a crucial identity: $R \, dT = V \, dP + P \, dV$ (for 1 mole). This equation links small changes in temperature, pressure, and volume.
The First Law of Thermodynamics, in the form used in this course, is $dQ = dU + P \, dV $, where $ P \, dV$ represents work done by the system. For an ideal monatomic gas, its internal energy $U = \frac{3}{2}RT$, leading to a change in internal energy $\Delta U = \frac{3}{2}R \, \Delta T$.
Joule’s experiments historically established the quantitative equivalence of heat and work. For instance, a $450 \, \text{m} $ waterfall warms by approximately $ 1^\circ\text{C} $ due to the conversion of potential energy into internal energy, following the relationship $ mgh = mc\Delta T$.
The Joule free expansion experiment demonstrated that for an ideal gas, internal energy $U$ depends only on temperature ($T$). In this experiment, both heat ($Q$) and work ($W$) are zero, so $\Delta U = 0$, leading to no observed temperature change ($\Delta T = 0$) despite changes in pressure and volume.
The setup for adiabatic processes begins with the First Law and the ideal gas internal energy, leading to the relation $C_V \, dT = -P \, dV $. Combining this with the ideal gas law also yields Mayer's relation, $ C_P = C_V + R $. The full derivation of adiabatic relations (like $ PV^\gamma = \text{constant}$) will be completed in the next lecture.
## Lecture 9: First Law Revisited
This lecture provides a bridge from our recent discussions on real gases and phase diagrams back to the foundational principles of classical thermodynamics. Our aim is to make the First Law of Thermodynamics operational, clearly distinguishing between state functions and processes. We'll learn how to calculate changes in internal energy for an ideal gas and lay the groundwork for understanding adiabatic relations, which we'll complete in the next session.
By the end of this lecture, you should be able to:
* Calculate changes in internal energy from considerations of work and heat.
* Calculate heat capacities and temperature differences, and understand their connection.
* Understand the consequences of the Joule free expansion experiment.
* Connect microscopic and macroscopic interpretations of the First Law.
* Set up the initial properties for an adiabatic expansion, leading to the full $PV^\gamma$ result next time.

### 1) Recap: real-gas isotherms, the phase dome, and $T_C \sim \varepsilon/k$
We've previously explored how real gases deviate from the ideal gas law, $PV = RT$. For an ideal gas, an isotherm (a curve of constant temperature on a pressure-volume diagram) is a smooth hyperbola. However, real gases, particularly at low temperatures, exhibit more complex behaviour. Their isotherms can enter a two-phase region, showing a characteristic "loop" that physically represents the process of condensation, where gas and liquid coexist.
The failure of the ideal gas model stems from two main assumptions. Firstly, ideal gases are assumed to have no intermolecular forces, but in reality, attractive forces exist between molecules. These attractions reduce the measured pressure on the container walls, which we account for with the van der Waals $a/V^2$ term. Secondly, ideal gas particles are treated as point-like, having negligible size. Real molecules, however, occupy a finite volume, reducing the free space available for other molecules to move in. This is corrected by the van der Waals $nb$ term, where $b$ represents the excluded volume per mole.
As we increase the temperature, the distinctive "loop" on the real gas isotherm shrinks. At a specific temperature, known as the critical temperature ($T_C$), this loop vanishes, replaced by a single point of inflection. Above $T_C$, no amount of compression can liquefy the gas; the liquid and gas phases become indistinguishable, forming a supercritical fluid. Conceptually, at this critical point, the thermal energy $kT_C$ is just sufficient to overcome the intermolecular binding energy, $\varepsilon$. This provides a useful approximation for the critical temperature: $T_C \sim \frac{\varepsilon}{k}$. This link connects the macroscopic critical temperature to the microscopic bond strength, $\varepsilon$, which we inferred earlier from latent heats and neighbour counting.
Water exhibits an interesting anomaly in its phase diagram. Unlike most substances, its solid-liquid boundary line has a negative slope. This occurs because ice is less dense than liquid water. Consequently, increasing the pressure on ice can cause it to melt, which is contrary to the behaviour of most materials where increased pressure favours the denser solid phase.

### 2) Thermodynamic terms and state functions
To understand energy transformations, we often consider a model system: a gas confined in a cylinder with a frictionless piston. The walls of the cylinder can be either diathermal, allowing heat to flow through, or adiabatic, preventing any heat exchange. When such a system is in thermodynamic equilibrium, it means no macroscopic properties are changing. This encompasses mechanical equilibrium (no net forces, so the piston isn't moving), thermal equilibrium (no net heat flow), and chemical equilibrium (no net change in chemical composition).
The macroscopic state of a system is defined by its state variables, or state functions, such as pressure ($P$), volume ($V$), and temperature ($T$). The value of a state function depends only on the current state of the system, not on the path taken to reach that state. Any equation that relates these state variables is called an equation of state; the ideal gas law, $PV = RT$ (for one mole), is a familiar example.

### 3) Differential form of the ideal gas law
When analysing small changes in a system, it's useful to express the ideal gas law in its differential form. Starting with $PV = RT$ for one mole of an ideal gas, we can consider an infinitesimal change. Applying the product rule for differentiation to the left side, $d(PV) = P\,dV + V\,dP$. The right side simply becomes $d(RT) = R\,dT$. Equating these, we obtain:
$$ R\,dT = V\,dP + P\,dV $$
This identity is a cornerstone for calculations involving thermodynamic processes. It directly links small changes in temperature to the corresponding small changes in pressure and volume, allowing us to track how these state variables evolve during a process.

### 4) Internal energy $U$: what it is and what we can measure
Internal energy, denoted by $U$, represents the sum of all microscopic energies within a system. This includes the translational, rotational, and vibrational kinetic energies of its molecules, as well as the potential energies arising from intermolecular interactions (which are significant in real gases).
Crucially, $U$ is a state function, just like pressure, volume, and temperature. This means its value depends solely on the system's current state, not on the particular path or process used to reach that state. In contrast, heat ($Q$) and work ($W$) are not properties of a system's state; they are processes, representing energy in transit as it moves into or out of the system. Because $U$ is a state function, the change in internal energy, $\Delta U$, between any two states depends only on the initial and final states, making it path-independent. In practical terms, we can only measure changes in internal energy, $\Delta U$, rather than its absolute value. This path independence is formally expressed by the concept of an exact differential, meaning that $dU$ can be written as a function of state variables, such as $dU(V,P) = (\frac{\partial U}{\partial V})_P dV + (\frac{\partial U}{\partial P})_V dP$, and its integral is independent of the path taken.

### 5) The First Law: forms and sign convention used in this course
The First Law of Thermodynamics is a statement of the conservation of energy. It quantifies how the internal energy of a system changes when energy is transferred as heat or work. One common form of the law states that the change in internal energy ($\Delta U$) is the sum of the heat added to the system ($Q$) and the work done on the system ($W$):
$$ \Delta U = Q + W $$
In this course, for derivations involving infinitesimal changes, we will primarily use the differential form:
$$ dQ = dU + P\,dV $$
Here, $P\,dV$ represents the infinitesimal work done *by* the system during an expansion. Thus, this equation states that the heat added to a system ($dQ$) can either increase its internal energy ($dU$) or be used by the system to do work on its surroundings ($P\,dV$). It's important to be aware that different textbooks may use different sign conventions for heat and work. Always check the specific form being used. In our convention, work done *by* the system enters as a positive $P\,dV$ term on the right-hand side of the equation.
### 6) Worked example: $\Delta U$ for heating 1 mol monatomic ideal gas from 0°C to 100°C
Let's calculate the change in internal energy when one mole of a monatomic ideal gas is heated from $0^\circ\text{C}$ to $100^\circ\text{C}$. For a monatomic ideal gas, the internal energy $U$ arises solely from translational kinetic energy. For one mole, this is given by $U = \frac{3}{2}RT$. This means that for an ideal gas, the internal energy depends only on its temperature, $T$.
To find the change in internal energy, $\Delta U$, we first find the rate of change of $U$ with respect to $T$:
$$ \frac{dU}{dT} = \frac{3}{2}R $$
Now, to find the total change in internal energy over a temperature range $\Delta T$, we can separate variables and integrate:
$$ \int_{U_1}^{U_2} dU = \int_{T_1}^{T_2} \frac{3}{2}R\,dT $$
$$ \Delta U = \frac{3}{2}R\,\Delta T $$
Given that $\Delta T = 100\,\text{K}$ (from $0^\circ\text{C} = 273.15\,\text{K}$ to $100^\circ\text{C} = 373.15\,\text{K}$), and using the ideal gas constant $R \approx 8.314\,\text{J mol}^{-1}\text{K}^{-1}$:
$$ \Delta U = \frac{3}{2} \times 8.314\,\text{J mol}^{-1}\text{K}^{-1} \times 100\,\text{K} \approx 1.25 \times 10^3\,\text{J} $$
So, heating one mole of a monatomic ideal gas by $100\,\text{K}$ increases its internal energy by approximately $1250\,\text{J}$.

### 7) Joule’s experimental foundations: heat-work equivalence
James Prescott Joule conducted seminal experiments in the mid-19th century that established the quantitative equivalence between heat and mechanical work, laying a crucial foundation for the First Law of Thermodynamics.
In his famous paddle-wheel experiment, Joule used a falling weight to rotate a paddle wheel submerged in an insulated container of water. He measured the mechanical work done by the falling weight and observed a corresponding rise in the water's temperature. He then demonstrated that the same temperature increase could be produced by supplying an equivalent amount of heat. This direct, quantitative equivalence between mechanical work and heat provided strong experimental support for the idea that internal energy can be changed by either process, as captured by the First Law, $\Delta U = Q + W$.

Joule even took thermometers on his honeymoon to the Swiss Alps, where he attempted to measure the temperature change of water falling over a $450\,\text{m}$ waterfall. The principle is that the gravitational potential energy lost by the falling water is converted into internal energy, causing a temperature rise. We can estimate this temperature change by equating the potential energy to the thermal energy:
$$ mgh = mc_w\Delta T $$
where $m$ is the mass of water, $g$ is the acceleration due to gravity, $h$ is the height of the waterfall, and $c_w$ is the specific heat capacity of water. Rearranging to solve for $\Delta T$:
$$ \Delta T = \frac{gh}{c_w} $$
Using $g \approx 9.8\,\text{m s}^{-2}$, $h = 450\,\text{m}$, and $c_w \approx 4190\,\text{J kg}^{-1}\text{K}^{-1}$, we find:
$$ \Delta T = \frac{9.8\,\text{m s}^{-2} \times 450\,\text{m}}{4190\,\text{J kg}^{-1}\text{K}^{-1}} \approx 1.05\,\text{K} $$
Thus, a $450\,\text{m}$ waterfall would cause the water to warm by approximately $1^\circ\text{C}$. This back-of-the-envelope calculation illustrates the magnitude of the effect Joule was trying to measure.

### 8) Joule free expansion: what $U$ depends on for an ideal gas
Joule also performed a free expansion experiment to investigate how the internal energy of a gas depends on its volume. In this setup, a thermally insulated container is divided into two compartments by a valve. One compartment contains a gas, while the other is evacuated (a vacuum). When the valve is opened, the gas rapidly expands to fill both compartments.
For an ideal gas undergoing this process, no heat is exchanged with the surroundings because the container is thermally insulated ($Q = 0$). Furthermore, since the gas expands into a vacuum, it does no work against any external pressure ($W = 0$). According to the First Law of Thermodynamics, $\Delta U = Q + W$, which means $\Delta U = 0$ for this experiment. Experimentally, for gases that behave ideally, no temperature change ($\Delta T = 0$) is observed during a free expansion. Since the pressure ($P$) and volume ($V$) of the gas change, but its internal energy ($U$) and temperature ($T$) do not, this experiment demonstrates a crucial property of ideal gases: their internal energy $U$ depends only on temperature ($U = U(T)$), not on volume or pressure.
For a real gas, a small cooling effect is actually observed. This is because real gas molecules have attractive intermolecular forces. As the gas expands, its molecules move further apart, doing work against these attractive forces. This internal work consumes some of the gas's kinetic energy, leading to a slight decrease in its temperature.

### 9) Setting up adiabatic process relations (derivation to be completed next lecture)
We can now begin to set up the relationships for an ideal gas undergoing an adiabatic process, where no heat is exchanged with the surroundings. This derivation will be completed in the next lecture, but we can establish the initial steps.
We start with the First Law of Thermodynamics in its differential form:
$$ dQ = dU + P\,dV $$
For an adiabatic process, $dQ = 0$. For an ideal gas, we know that the change in internal energy is given by $dU = C_V\,dT$, where $C_V$ is the molar heat capacity at constant volume. Substituting these into the First Law:
$$ 0 = C_V\,dT + P\,dV $$
This can be rearranged to:
$$ C_V\,dT = -P\,dV $$
We also use the differential form of the ideal gas law for one mole, $R\,dT = V\,dP + P\,dV$. From this, we can derive Mayer's relation. At constant pressure ($dP=0$), the ideal gas law simplifies to $R\,dT = P\,dV$. Substituting this into the First Law (at constant pressure, $dQ = C_P\,dT$ and $dU = C_V\,dT$):
$$ C_P\,dT = C_V\,dT + R\,dT $$
Dividing by $dT$, we get Mayer's relation:
$$ C_P = C_V + R $$
These ingredients, including $C_V\,dT = -P\,dV$ and Mayer's relation, allow us to derive the full adiabatic relations linking pressure, volume, and temperature for ideal gases, such as $PV^\gamma = \text{constant}$ (where $\gamma = C_P/C_V$). However, the complete derivation of these results will be covered in the next lecture.

### Slides present but not covered this lecture (for clarity)
Please note that while the slide deck for this lecture may contain the final $PV^\gamma$ result and its integration steps (often boxed on a slide), this specific content was not covered in this session. The full derivation of these adiabatic relations will be completed in the next lecture.
### Key takeaways
Real gases deviate from the ideal gas law ($PV = RT$) because their molecules have finite size and experience intermolecular attractions. These effects give rise to the two-phase "dome" on P-V diagrams and the concept of a critical point, where the distinction between liquid and gas vanishes. The critical temperature ($T_C$) is physically linked to the microscopic intermolecular separation energy ($\varepsilon$) by the approximate relationship $kT_C \sim \varepsilon$.
A system's macroscopic state is defined by state variables like pressure ($P$), volume ($V$), and temperature ($T$). Internal energy ($U$) is also a state function, meaning its value depends only on the system's current state. In contrast, heat ($Q$) and work ($W$) are processes, representing energy in transit, not properties of the state itself.
For infinitesimal changes in an ideal gas, the differential form of the ideal gas law is a crucial identity: $R\,dT = V\,dP + P\,dV$ (for 1 mole). This equation links small changes in temperature, pressure, and volume.
The First Law of Thermodynamics, in the form used in this course, is $dQ = dU + P\,dV$, where $P\,dV$ represents work done *by* the system. For an ideal monatomic gas, its internal energy $U = \frac{3}{2}RT$, leading to a change in internal energy $\Delta U = \frac{3}{2}R\,\Delta T$.
Joule’s experiments historically established the quantitative equivalence of heat and work. For instance, a $450\,\text{m}$ waterfall warms by approximately $1^\circ\text{C}$ due to the conversion of potential energy into internal energy, following the relationship $mgh = mc\Delta T$.
The Joule free expansion experiment demonstrated that for an ideal gas, internal energy $U$ depends only on temperature ($T$). In this experiment, both heat ($Q$) and work ($W$) are zero, so $\Delta U = 0$, leading to no observed temperature change ($\Delta T = 0$) despite changes in pressure and volume.
The setup for adiabatic processes begins with the First Law and the ideal gas internal energy, leading to the relation $C_V\,dT = -P\,dV$. Combining this with the ideal gas law also yields Mayer's relation, $C_P = C_V + R$. The full derivation of adiabatic relations (like $PV^\gamma = \text{constant}$) will be completed in the next lecture.